Definición:
 Según la Organización Mundial de la Salud OMS: «Cáncer» es un término genérico que designa un amplio grupo de
enfermedades que pueden afectar a cualquier parte del organismo; también se habla
de «tumores malignos» o «neoplasias malignas». (1)
Según la Organización Mundial de la Salud OMS: «Cáncer» es un término genérico que designa un amplio grupo de
enfermedades que pueden afectar a cualquier parte del organismo; también se habla
de «tumores malignos» o «neoplasias malignas». (1)
Según el Instituto
Nacional del Cáncer NIH: Cáncer es el nombre que se da a un conjunto de
enfermedades relacionadas. En todos los tipos de cáncer, algunas de las células
del cuerpo empiezan a dividirse sin detenerse y se diseminan a los tejidos del
derredor.(2)

En general podemos decir que el cáncer se
origina por el crecimiento celular incontrolado en algunaparte del cuerpo. La
trasformación de las células normales en célulascancerosas surge como
consecuencia del daño del DNA, dando lugara células incapaces de controlar su
crecimiento y división. (4)
 La malignidad del
cáncer es variable, según la agresividad de sus células y demás características
biológicas de cada tipo tumoral. En general, el comportamiento de las células
cancerosas se caracteriza por carecer del control reproductivo que requiere su
función original, perdiendo sus características primitivas y adquiriendo otras
que no les corresponden, como la capacidad de invadir de forma progresiva y por
distintas vías a órganos próximos (metástasis),
con crecimiento y división más allá de los límites normales del órgano,
diseminándose por el organismo fundamentalmente a través del sistema linfático o el sistema circulatorio, y ocasionando el
crecimiento de nuevos tumoresen otras partes del cuerpo alejadas de la localización
original. (5)
La malignidad del
cáncer es variable, según la agresividad de sus células y demás características
biológicas de cada tipo tumoral. En general, el comportamiento de las células
cancerosas se caracteriza por carecer del control reproductivo que requiere su
función original, perdiendo sus características primitivas y adquiriendo otras
que no les corresponden, como la capacidad de invadir de forma progresiva y por
distintas vías a órganos próximos (metástasis),
con crecimiento y división más allá de los límites normales del órgano,
diseminándose por el organismo fundamentalmente a través del sistema linfático o el sistema circulatorio, y ocasionando el
crecimiento de nuevos tumoresen otras partes del cuerpo alejadas de la localización
original. (5)

 El cáncer comienza
en una célula. La transformación de una célula normal en tumoral es un proceso
multifásico y suele consistir en la progresión de una lesión precancerosa a un
tumor maligno. Estas alteraciones son el resultado de la interacción entre los
factores genéticos del paciente y tres categorías de agentes externos
establecidos por la OMS a través de su Centro Internacional de Investigaciones
sobre el Cáncer, los agentes cancerígenos se clasifican en: (1)
El cáncer comienza
en una célula. La transformación de una célula normal en tumoral es un proceso
multifásico y suele consistir en la progresión de una lesión precancerosa a un
tumor maligno. Estas alteraciones son el resultado de la interacción entre los
factores genéticos del paciente y tres categorías de agentes externos
establecidos por la OMS a través de su Centro Internacional de Investigaciones
sobre el Cáncer, los agentes cancerígenos se clasifican en: (1)


La malignidad del
cáncer es variable, según la agresividad de sus células y demás características
biológicas de cada tipo tumoral. En general, el comportamiento de las células
cancerosas se caracteriza por carecer del control reproductivo que requiere su
función original, perdiendo sus características primitivas y adquiriendo otras
que no les corresponden, como la capacidad de invadir de forma progresiva y por
distintas vías a órganos próximos (metástasis),
con crecimiento y división más allá de los límites normales del órgano,
diseminándose por el organismo fundamentalmente a través del sistema linfático o el sistema circulatorio, y ocasionando el
crecimiento de nuevos tumoresen otras partes del cuerpo alejadas de la localización
original. (5)
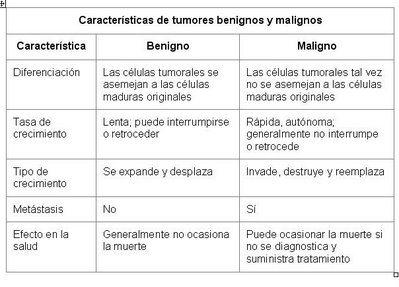
Existen diferencias entre los tumores benignos
y los malignos:
Tabla 1. Diferencias entre los Tumores (5)

Los tumores malignos se puede clasificar
según:
- · Lugar de origen, ej. Mama, pulmón, colon, etc.
- · Tejido o célula del que procede; (6)
o
Tejidos mesenquimatosos sólidos = Sarcoma
o
Células epiteliales = Carcinoma
o
Células escamosas = Carcinoma epidermoide
o
Células tumorales similares al
epitelio escamoso estratificado = Adenocarcinoma
o
Células formadoras de sangre = Leucemia
o
Linfocitos o precursores = Linfoma
- · Extensión:
o
Sistema TNM: (5)

o
Estadios del tumor;
§ Estadio I: pequeños, localizados y habitualmente curables.
§ Estadio II y III: localmente avanzados y/o con afectación de los
ganglios linfáticos locales.
§ Estadio IV: metastáticos y en la mayoría de casos inoperables.
Estos
estadios se definen con gran precisión, siendo diferente para cada tipo de
cáncer.
El estado general de
los pacientes con cáncer se puede medir mediantes escalas internacionales que
evalúan de forma estándar la capacidad del paciente para realizar sus tareas
rutinarias.
La puntuación de la
escala de rendimiento de Karnofsky (KPS)
oscila entre 0 y 100. Una puntuación más alta significa que el paciente tiene
mejor capacidad de realizar las actividades cotidianas. La KPS se utiliza para
determinar el pronóstico del paciente, medir los cambios en la capacidad del
paciente para funcionar o decidir si un paciente puede ser incluido en un
estudio clínico e incluso para decidir si un paciente es o no candidato a
recibir un tratamiento con quimioterapia. (5)
Tabla 2. Escala de Rendimiento de Karnofsky (KPS) (5)
Existen otras escalas similares como la escala
ECOG, donde lapuntuación oscila entre 0 y 5. La puntuación más alta significa
que elpaciente tiene mejor capacidad para realizar actividades de la
vidadiaria. (5)
Tabla
3. Escala ECOG (5)
GRADO
|
ECOG
|
0
|
Asintomático. Capaz de llevar a cabo actividad normal
|
1
|
Sintomático. Restricción en su actividad física diaria
pero totalmente ambulatorio
|
2
|
Sintomático. Encamado <50% del día. Capaz de cuidarse pero incapaz
de llevar a cabo actividad física o laboral.
|
3
|
Encamado >50% del día. Requiere atención ocasional.
|
4
|
Encamado el 100% del día. Invalidez severa
|
5
|
Muerto
|
CAUSAS
DEL CÁNCER:
El cáncer comienza
en una célula. La transformación de una célula normal en tumoral es un proceso
multifásico y suele consistir en la progresión de una lesión precancerosa a un
tumor maligno. Estas alteraciones son el resultado de la interacción entre los
factores genéticos del paciente y tres categorías de agentes externos
establecidos por la OMS a través de su Centro Internacional de Investigaciones
sobre el Cáncer, los agentes cancerígenos se clasifican en: (1)
·
Carcinógenos físicos, como las
radiaciones ultravioleta e ionizantes;
· Carcinógenos químicos, como los
asbestos, los componentes del humo de tabaco, las aflatoxinas (contaminantes de
los alimentos) o el arsénico (contaminante del agua de bebida);
· Carcinógenos biológicos, como las
infecciones causadas por determinados virus, bacterias o parásitos. (1)
El envejecimiento
es otro factor fundamental en la aparición del cáncer. La incidencia de esta
enfermedad aumenta muchísimo con la edad, muy probablemente porque se van
acumulando factores de riesgo de determinados tipos de cáncer. La acumulación
general de factores de riesgo se combina con la tendencia que tienen los
mecanismos de reparación celular a perder eficacia con la edad. (1)
FACTORES DE RIESGO QUE PUEDEN DESARROLLAR CÁNCER
El consumo de
tabaco y alcohol, la dieta malsana y la inactividad física son los principales
factores de riesgo de cáncer en todo el mundo. Algunas infecciones crónicas
también constituyen factores de riesgo, y son más importantes en los países de
ingresos medios y bajos. (1)
Los virus de las
hepatitis B (VHB) y C (VHC) y algunos tipos de papilomavirus humanos (PVH)
aumentan el riesgo de cáncer de hígado y cuello uterino, respectivamente. La
infección por el VIH también aumenta considerablemente el riesgo de algunos
cánceres, como los del cuello uterino. (1)

BIOLOGÍA CELULAR DEL CÁNCER
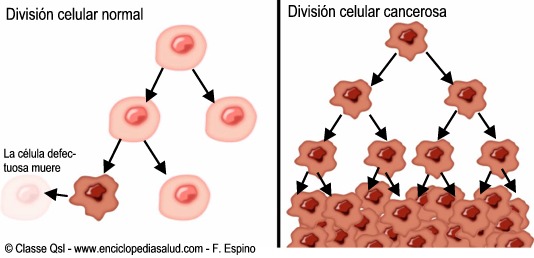
Existen
alteraciones celulares esenciales para la transformación maligna de una célula,
es decir la reproducción de una célula cancerosa y provocar cáncer:
·
Gran división celular, aunque no
siempre es indicativo de que un tumor es maligno o que el tejido sea neoplásico
·
Perdida de la diferenciación
celular o anaplasia, acciones que se ve asociada a otras alteraciones
morfológicas:
o
Pleomorfismo; es decir alteración
en su tamaño y forma.
o Morfología nuclear anómala; núcleo
extremadamente grandes, relación núcleo-citoplasma desproporcionada 1:1.
o
Figuras mitóticas atípicas y
grotescas, en ocasiones husos tripolares, tetrapolares o multipolares son un
rasgo que denota malignidad.
o
Pérdida de la polaridad celular.
o
Áreas de necrosis isquémica en
tumores malignos de rápido crecimiento
·
Alteraciones displásicas o
metaplásicas
·
Invasión local
·
Metástasis (6)
{kind=link}
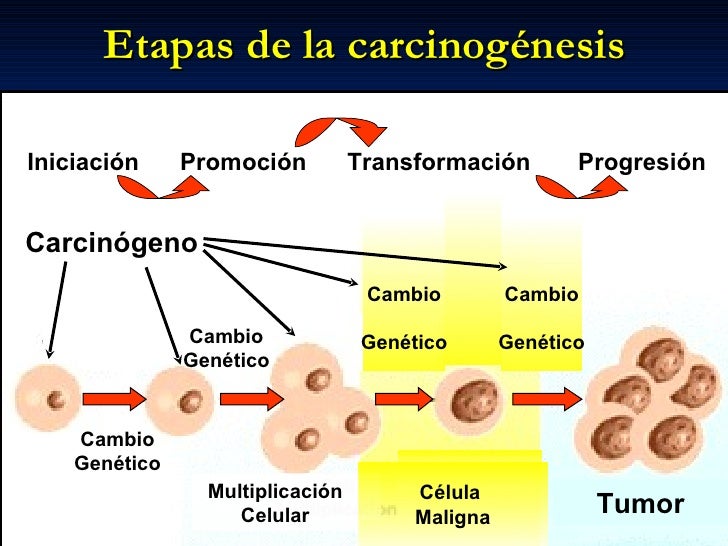
El proceso por el
cual las mutaciones producidas en el DNA de célulassanas llevan a la aparición
de células cancerosas se denominacarcinogénesis, siendo de duración variable,
según el tipo de cáncer,aunque de manera general podemos reconocer cuatro fases
biendiferenciadas:
1.
Inducción o iniciación:
Aparecen las mutaciones del DNAque dotan a la célula de lascaracterísticas
propias de lacélula cancerosa: división incontrolada, capacidad deinvasión
local y de diseminación a distancia.
2.
Cáncer “in situ”: Aumento del
número de célulascancerosas en el órgano en el que se origina. Se vagenerando
el tumor primario.
3.
Invasión local: Extensión del
tumor primario a lasestructuras vecinas, invadiéndolas. Aparición de síntomas.
4.
Invasión a distancia o
metastatización: las célulascancerosas acceden a cavidades y superficies
corporales, al torrente sanguíneo o linfáticodiseminándose a órganos a
distancia, y originando tumoressecundarios denominados metástasis.
A lo largo de cada
una de estas fases las células van acumulandomutaciones que hacen que pierdan
sus características alterando su funcionamiento.
MECANISMOS
PROTOONCOGENES
Se pueden clasificar en la función de la proteína codificada:


Ejemplos:
PATOGENIA DEL RETINOBLASTOMA.

REFERENCIAS BIBLIOGRAFICAS
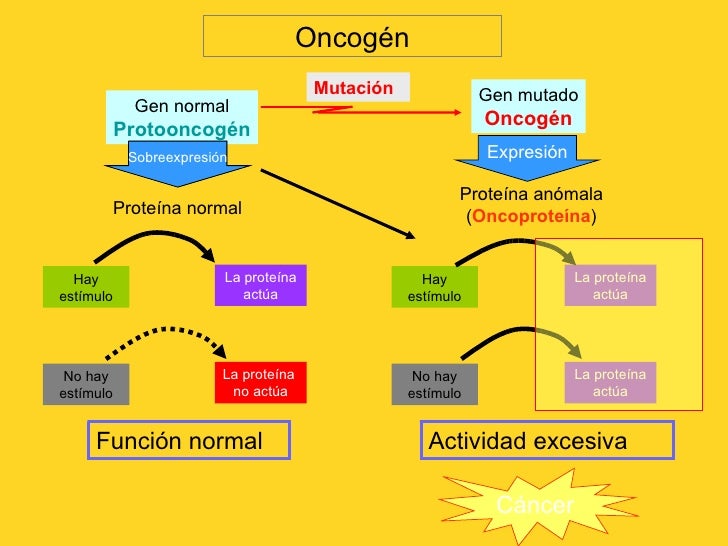
PROTOONCOGENES
Los protooncogenes, son genes que son
normales, pero estos pueden llegar a convertirse en oncogenes por: mutaciones
puntuales, reordenamientos cromosómicos y por la amplificación de los genes.
Entre sus funciones están, transducción de señales, ejecución de señales
mitogénicas, las cuales actúan en puntos estratégicos del crecimiento y
diferenciación celular.
Definición
|
Causa
|
|
Mutaciones Puntuales
|
15 % de todos los tumores humanos tienen oncogenes
H-ras o K-ras. Se puede dar por la exposición a sustancias químicas que
pueden llegar a producir cáncer.
|
Sustitución de un par de bases por otro par en una
secuencia de ADN.
|
Reordenamientos cromosómicos
|
Colocación de genes en las proximidades de los
promotores/potenciadores en los loci de receptores de las células T o
inmunoglobulinas.
Fusión de gen con las nuevas secuencias genéticas.
|
Inserción del ADN del virus en el genoma del
huésped.
|
Amplificación de genes
|
Reduplicación de los protooncogenes, que llevan a
inducir un aumento de su expresión o la actividad.
|
Una de las copias lleva a multiplicarse miles de
veces.
|
ONCOGENES
Son genes que han sufrido cambio, las cuales llegan a promover el
crecimiento celular autónomo en las células cancerosas, por una ausencia de señales
promotoras del crecimiento normalmente(6)
Estos codifican una proteína anómala, la cual
se mantiene activa independiente de las señales reguladores y convierte a la
célula en una célula tumoral.
Se clasifican en el lugar de acción:
Extracelular
|
Membrana
|
Citoplasma
|
Núcleo
|
Fuerzan a la
célula a producir un exceso de factores de crecimiento, además pueden activar
la proliferación de las células que los produjeron.
|
Se producen versiones oncogénicas de receptores
celulares para factores de crecimiento.
|
Se generan versiones oncogénicas de proteínas del
citoplasma que se mantienen siempre activas.
|
Se producen versiones oncogénicas de factores de
transcripción o secuencias asociadas que funcionan en todo momento.
|
Se pueden clasificar en la función de la proteína codificada:
LA PROTEINA AFECTADA PUEDE SER
|
Proteínas G
|
Factores de
crecimiento
|
Proteínas quinasas
de serina- treonina y tirosina
|
Factores de
transcripción nuclear
|
Productos que
afectan a la apoptosis
|
Mecanismos
De Transformación:
|
||||
Métodos
cualitativos
|
Inserción de
promotor viral
|
Translocación
o reordenación cromosómica,
|
Amplificación
|
Hipometilación
|
Sustitución de una base
nitrogenada en el ADN de un gen, provocan un cambio estructural en la
proteína sintetizada por ese gen, alterándose su función.
|
Genes celulares que se han incorporado al genoma del virus en una
infección previa, y que son transmitidos a su genoma lo cual afectaría
después a otros organismos.
|
Es el cambio de localización de una porción
cromosómica, con los genes que lleva incorporados a otra ubicación distinta
dentro del mismo cromosoma o de otro.
|
Es el aumento del número de copias del mismo
protooncogenes del genoma
|
Entre el 2 y 7% de residuos de citosina en el ADN
están metilados.
|
GENES SUPRESORES
Son los grupos de genes los cuales tienen
la capacidad de inhibir la proliferación celular, li hace porque bloquean la
actividad de oncogenes y de los productos de oncogenes. El fallo en estos genes
debe ser expresado en ambos alelos, para llegar a provocar una alteración
fenotípica que comprometa la fisiología de la célula o puede llegar a ser
germinal y darse de manera esporádica.
Los genes supresores de tumores guardianes
codifican, los reguladores de diversos puntos de control del ciclo celular y
los mediadores de la muerte celular programada, donde a esto al mecanismo por
el cual actúan. Los genes cuidadores o de mantenimiento son las proteínas
responsables de la detección y la reparación de las mutaciones, las proteínas
implicadas en las disyunciones cromosómicas normales durante la mitosis y
componentes del dispositivo de muerte celular programada.
Los genes más conocidos son: p53,
retinoblastoma, DCC, MDD, APC, NF1, NF2 y WT-1
MODO DE ACCION DE LOS ONCOGENES EN TUMORES HUMANOS ASOCIADOS
Los genes que fomentan el crecimiento celular autónomo de las
células cancerosas se denominan oncogenes, y los genes celulares
correspondientes no mutados se denominan protooncogenes.
Los oncogenes se generan por mutaciones de los protooncogenes y
codifican proteínas denominadas oncoproteínas, que inducen el crecimiento
celular sin que exista la señal normal correspondiente.
En condiciones fisiológicas, las vías de señalización de los factores
de crecimiento se resumen en estas etapas:
1.
Unión de un factor de
crecimiento al receptor específico
2.
Activación pasajera y limitada
del receptor del factor de crecimiento, que, a su vez activa proteínas
citoplasmáticas traductoras de la señal
3.
Transmisión de la señal
traducida al núcleo a través de nuevas proteínas y segundos mensajeros, o de
una cascada de moléculas traductoras de la señal
4.
Inducción y activación de los
factores reguladores nucleares que inician la transcripción del ADN
5.
Expresión de factores que
fomentan la entrada en la progresión de la célula dentro del ciclo, la división
celular
6.
Se producen cambios en la
expresión de otros genes que respaldan la supervivencia celular y las
modificaciones metabólicas necesarias para un crecimiento óptimo
a. Factores de
crecimiento: las células normales, para proliferar,
necesitan el estímulo de los factores de crecimiento. La mayoría de los
factores de crecimiento solubles son sintetizados por un único tipo celular y
actúan sobre la célula vecina estimulando la proliferación (acción paracrina).
Sin embargo, ciertas células cancerosas adquieren la capacidad para sintetizar
estos mismos factores de crecimiento, a los que responden creando un bucle
autocrino. Por ejemplo, muchos tumores cerebrales denominados glioblastomas,
expresan el factor de crecimiento derivado de las plaquetas (PDGF).(6)
b. Receptores de los
factores de crecimiento: un número elevado de
oncogenes codifican receptores de factores de crecimiento, entre los que
destacan los receptores de tirosina cinasa. Estos receptores de tirosina cinasa
se pueden activar en el tumor por diversos mecanismos como: mutaciones
puntuales, reordenamientos génicos y amplificaciones génicas.(6)
Tabla 7-5 oncogenes selectos, modo de
activación y tumores humanos asociados.
c. Proteinas que participan en la transduccion de señales
PROTEINAS G ASOCIADAS A MEMEBRANA
Las proteínas RAS pertenece a las familia de proteínas G asociadas a
membrana, cuando están unidas a GTP, se encuentra en un estado activado,
mientras estas se encuentre unido a GDP se encuentran inactivas. El intercambio
de GDP por GTP lo determinan los
factores de crecimiento al estimular losreceptores de tirosina cinasa. La activación de las RAS es transitoria gracias a su actividad
intrínseca GTPasa,que aumentando su funcióninactivada por las proteínas
activadoras de las GTPasa (GAP), se dice que la aceleramás de 1000 veces. Podríamos
decir que las GAP impiden una actividad
descontrolada de las RAS al participar en su inactivación. Una RAS activada
favorece la transducción de señales que estimula las ramas de MAPK Y PI3K/AKT, luego
estas fosforilan y activan genes favorecedores del crecimiento celular rápido
por medio de efectores citoplasmáticos y
diversos factores de transcripción.(6)
Las mutaciones de la familia de los RAS constituyen la anomalíamás
común de los protooncogenes en los tumores humanos, estas mutaciones hacen que
las RAS se queden en forma activada es
decir, favoreciendo las señales de crecimientocelular.
Dentro de las RAS mutadas se encuentras los siguientes: KRAS, HRAS,
NRAS, GNAQ, GNAS. Estas mutaciones puntuales se encontraron en ell 90% de los adenocarcinomas pancreáticos y
colangiocarcinomas, 50% en cáncer de colon, endometrio y tiroides; y en el 30%
de leucemias mieloides y adenocarcinomas de pulmón.(6)
PROTEINAS CINASA
El protooncogen BRAF miembro de la familia RAF, es una proteína
cinasa de serina/ treonina que encabeza la cascada de la familia de las MAPK que es una cinasa de serina/ treonina,
una mutación de BRAF termina estimulando factores de trascripción por medio de
MAPK que en última instancia confiere un aumento de crecimiento celular. Estas
mutaciones se han encontrado en casi todas las leucemias de células peludas, en
80% de nevos benignos y en más del 60% de los melanomas.
La proteína PI3K es un heterodimero que tiene una subunidad
catalítica y una reguladora, activa una cascada de cinasa de serina/treonina
como AKT, punto esencial en laseñalización, pues por medio de mTOR activa la
síntesis de proteínas y lípidos, estos dos juntos favorecen el crecimiento
celular. PTEN frena la actividad de PI3K, por lo que se conoce como gen
supresor de tumores, pierde su función cuando exite un silenciador epigenético
o una mutación,cuando existe esta última,
afectan mayormente a la subunidad catalítica de la PI3K aumentando su actividad
enzimática.
TIROCINAS CINASAS NO ASOCIADAS A RECEPTORES
Las alteraciones en tirosinas cinasas no asociadas a receptores se
ubican normalmente en el citoplasma y núcleo, sin embargo activan las mismas
vías de señalización que los receptores de tirosina cinasa. Habitualmente
sufren de una translocación o reordenamientos cromosómicos que generan
genes de fusión, estos últimos, codifican tirosinas cinasas con actividad constitucional.
Entre estas tenemos ABL: en la leucemia
mieloide crónica y en algunas leucemias linfoblasticas agudas, este gen se transloca
del cromosoma 9 hasta el cromosoma 22, donde se fusiona con el gen BCR, codificando
un a tirocina cinasa BCR-ABL oncogéna y constitucionalmente activa.
Otra mutación presentada en este grupo es la anulación de dominios
reguladores negativos que mantienen controlada la actividad enzimática, por
ejemplo JAK2 que interviene en
JAK/STAT: traduce señales mitógenas del factor de crecimiento y de receptores
citocinicos que carecen de actividad tirocina cinasa, su activación altera expresión
de genes diana que ligan los factores de trascripción STAT.(6)
d. Proteinas reguladoras nucleares
Las señales de las vías de transducción convergen en el núcleo,
finalmente las señales mitógenas desreguladas estimulan continua e inapropiadamente factores de
transcripción nucleares de genes
favoreciendo el crecimiento celular.
MYC: casi todas las vías de señalización que favorecen el crecimiento
convergen aquí, es el que mas participa en los tumores humanos, es expresado en
casi todas las células eucariotas, son inducidos por RAS/MAPK, , en concentraciones normales se encuentra
reguladas a través de la transcripción traducción y estabilidad de la proteína,
activa la expresión de muchos genes que contribuyen al crecimiento celular,
regula el alza de expresión de la telomerasa, es uno de
los numerosos factores de transcripción que actúa en forma conjunta para
reprogramar célulassomáticas hacia células madre pluripotenciales. Una
desregulación de crecimiento celular ya sea por polimorfismos, alteración
genética del propio MYC o una translocación, amplificación o aumento de
concentración de MYCP contribuyen a mecanismos en elcáncer.(6)
e. Proteinas reguladores del Ciclo Celular
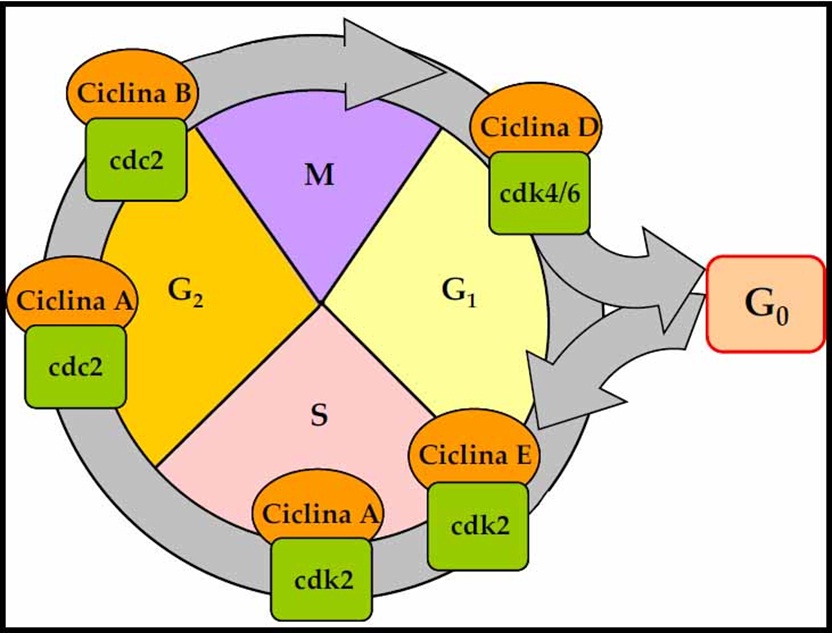
Existen dos puntos de control
en el ciclo celular, una se encuentra en G1/S, los defectos en este
punto determina crecimiento sin regulador adecuado y un fenotipo mutador
facilitando la aparición y progresión del cáncer por eso es elmás importante; y otra en G2/M.
Ambos puntos de control están dados por un equilibrio entre sensores de daño
de ADN que mandan señales hasta que ocurre apoptosis si el daño no se repara; y
también por factores que favorecen o suprimen el crecimiento. Entre las
proteínas que participan en este último proceso , se menciona a las cinasas
dependientes de ciclinas(CDK): se activan por unión a ciclinas, fosforilan
proteínas diana cruciales que conducen células a través del ciclo celular como
RB.(6)
Mutaciones adquiridas como
translocaciones cromosómicas y amplificación
génica de CDK hace que haya una
regulación anómala que favorece la velocidad del crecimiento tumoral en la
transición G1/S.El inhibidor de CDK (CDKI) silencia las CDK y ejercen control negativo
en el ciclo celular.
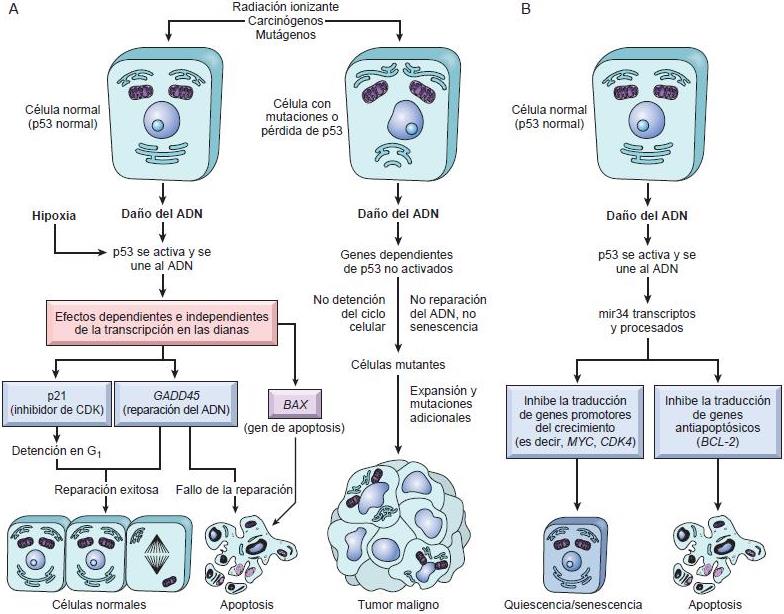
Los dos genes supresores de tumores más importantes P53 y
RB, codifican ambas proteínas que inhiben progresión G1/S.
P53 produce apoptosis y una parada del ciclo celular se necesita en
G1/S y es componente esencial en punto de control G2/M.
RB en forma de bolsa interacciona con factores de transcripción que
regula la diferenciación.
MODO DE ACCION DE LOS GENES SUPRESORES DE TUMORES HUMANOS
Un gen supresor tumoral es un gen que reduce la probabilidad de que
una célula en
un organismo multicelular se transforme en una célula cancerígena. Los
genes supresores de tumores se encuentran en las células normales y generalmente inhiben la
proliferación celular excesiva. Una mutación o una deleción de un gen supresor tumoral, aumentará la probabilidad de que se
produzca un tumor, al perder su función. De esa manera, un gen supresor tumoral
alterado es similar a un oncogén.
En las células normales, las proteínas codificadas por los genes
supresores de tumores detienen la progresión del ciclo celular en respuesta a
un daño en el ADN o a señales de supresión del crecimiento provenientes del
medio extracelular. Cuando los genes supresores de tumores estan mutados o son
inactivos, las células no pueden responder normalmente a los puntos de control
del ciclo celular, o son incapaces de realizar la muerte celular programada si
el daño del ADN es demasiado importante.(6)
- RB: la gobernadora de la proliferacion. La RB, es un regulador negativo esencial de la transicion del ciclo celular G1/S, sufre una inactivacion directa o indirecta en la mayoria de los cánceres humanos. La RB controla también la diferenciacion celular. Se encuentra en un estado hipofoforilado activo en las células quiescentes y en un estado hiperfosforilado inactivo en las que atraviesan la transición del ciclo celular G1 /S
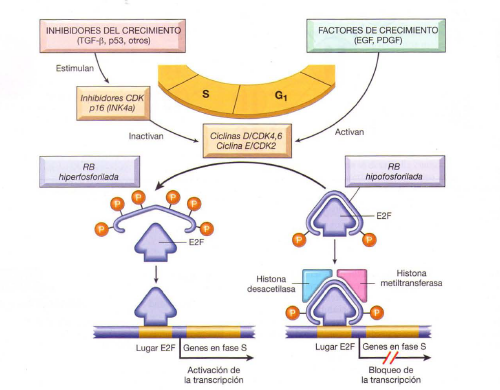
- p53: el guardián del genoma. El TP53, un gen supresor de tumors que regula la progresión del ciclo celular, la repación del ADN, la senecesncia celular y la apoptosis, es el gen que más veces muta en los cánceres humanos. Una vez activada, la p53 impide la transformación neoplásica al inducir una parada transitoria del ciclo celular, la senescencia ( parada permanente del ciclo celular) o la muerte celular programada (6) .
PATOGENIA DEL RETINOBLASTOMA.
El retinoblastoma
(RB) es un tumor embrionario de origen retiniano que se presenta generalmente
en niños menores de 5 años. Su incidencia es de 1:15-20.000 recién nacidos
vivos y su etiología esporádica en el 60% de los casos o hereditaria, en el
40%. Dejado a su evolución, el retinoblastoma es casi siempre fatal y un
retraso en su tratamiento suele suponer un pronóstico visual precario, de ahí
la transcendencia del diagnóstico precoz. El gen responsable, el RB1, está
localizado en el cromosoma 13 y actúa de forma dominante; es decir, en células
donde las dos copias del gen estén dañadas. El 35-40% de los pacientes
corresponden a casos hereditarios que son portadores de una mutación germinal.
De ellos, más de dos tercios representan nuevas mutaciones (“mutación de novo”)
sin historia familiar previa.
.
RETINOBLASTOMA
MANIFESTACIONES
CLINICAS
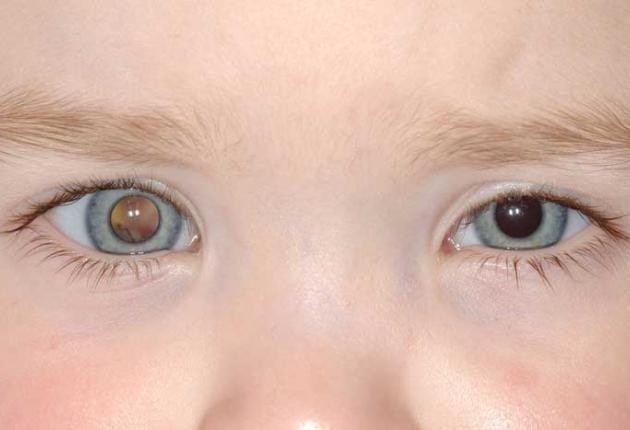
1. El
retinoblastoma se presenta clínicamente con leucocoria y estrabismo y más
raramente con glaucoma, celulitis orbital, uveitis y hemorragia vítrea. El 60%
de los RB son unilaterales con una edad media al diagnóstico de 24 meses,
mientras que los RB bilaterales son menos frecuentes, suelen ser multifocales y
de más temprana edad de comienzo (15 meses). En la mayoría de los niños con
tumores bilaterales, ambos ojos están afectados al diagnóstico. Sólo en algunos
casos de RB unilateral se desarrolla un tumor contralateral más tarde. Las
características de inicio precoz, bilateralidad y multifocalidad apuntan a un
probable origen hereditario del proceso.
2.- Los retinocitomas
o retinomas son tumores benignos resultantes de la regresión espontánea de
retinoblastoma y que raramente pueden ser descubiertos en la edad adulta como
lesión residual de un RB que cursó de forma subclínica en la infancia.
3.- Otros tumores
asociados: Los portadores de la mutación germinal en RB1 tienen un exceso de
riesgo de desarrollar otros tumores. La presencia de tumores extraoculares
llamados segundos tumores primarios, se manifiesta en la adolescencia o al
inicio de la edad adulta, siendo el tiempo medio de aparición del segundo tumor
10,4-13 años. El riesgo de padecerlos se incrementa en más de un 50% en
pacientes que han recibido radiación externa.
GENÉTICA DEL
RETINOBLASTOMA.
El retinoblastoma
está causado por mutaciones en el gen RB1. La mayoría de las familias con RB
hereditario presentan un patrón de transmisión autosómico dominante con
penetrancia casi completa y alta expresividad. Sin embargo, existen algunas
familias con característica baja penetrancia en las que se sugiere que las
mutaciones en RB1 dan lugar a menor cantidad de una proteína normal (mutaciones
Clase I) o a una proteína mutante parcialmente funcional (mutaciones Clase II). (7)
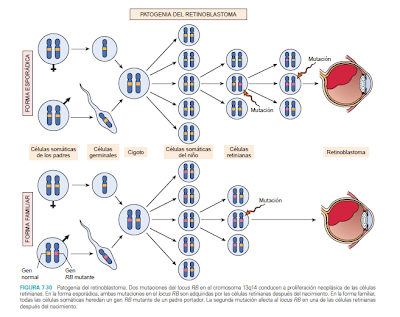
La carcinogénesis
del retinoblastoma sigue el modelo de las dos mutaciones o hipotésis de
Knudson, enunciado por primera vez para explicar la alta tasa de mutaciones de
esta enfermedad y según el cual los casos esporádicos son debidos a dos
mutaciones somáticas en una misma célula, mientras que los heredados ocurren en
personas susceptibles que ya son portadoras de la primera mutación o mutación
germinal.
El gen RB1, que se
comporta como dominante, es en realidad recesivo. Es decir, sólo se produce la
enfermedad cuando está en homo o hemicigosis. Este tipo de genes son
denominados genes reguladores o supresores de tumor porque, al contrario de los
oncogenes, su acción es necesaria para que el tumor no se desarrolle. Es por
este motivo por el que reciben el nombre de antioncogenes. La alta incidencia
de segundos tumores primarios en esta patología sugiere que este gen juega un
papel importante en la etiología de otros tumores primarios.(7)
El RB1 contiene 27
exones transcritos en un ARN de 4,7 kb. Codifica para una proteina nuclear que
controla la división de las células de la retina, regulando la transición de G1
a S. Se han descrito más de 200 mutaciones distintas. El 80-85% de ellas
resultan en una prematura terminacion del codon. El 80% de las mutaciones de
novo son de origen paterno, sugiriéndose que la mutación del RB1 es más frecuente
en la espermatogenesis.(7)
1.
Organización Mundial de la
Salud OMS. Cáncer [en línea] Febrero 2015 [citado 21 Ago 2016] Disponible en: http://www.who.int/mediacentre/factsheets/fs297/es/
2.
Instituto Nacional del Cáncer
NIH. ¿Qué es el cáncer? [en línea] 2016 [citado 21 Ago 2016] Disponible en:http://www.cancer.gov/espanol/cancer/naturaleza/que-es
3.
Sociedad Americana del Cáncer. Aspectos básicos sobre
el cáncer [en línea] 2015 [citado 21 Ago 2016] Disponible en:http://www.cancer.org/espanol/cancer/aspectosbasicossobreelcancer/index
4.
Fundación para la Excelencia y
la calidad de la Oncología ECO. Generalidades en Oncología [en línea] 2016
[citado 21 Ago 2016] Disponible en:http://www.fundacioneco.es/wp-content/uploads/2014/04/1.Generalidades.pdf
5.
Universidad de Cantabria UC.
Generalidades y tratamiento del cáncer [en línea] 2016 [citado 21 Ago 2016]
Disponible en:http://ocw.unican.es/ciencias-de-la-salud/enfermeria-clinica-i-2011/material-de-clase/bloque-i/Tema%201.4%20Generalidades%20y%20tratamiento%20del%20cancer.pdf
1.
Kumar, Vinay y Col. Patología
Estructural y Funcional 9ª. Edición. Editorial Elsevier Saunders España 2015.
(impreso en China) Neoplasias, capítulo 7,pag. 266-274.
7. Alonso A, Moreno S. GUÍA DE MANEJO DE RETINOBLASTOMA [en linea] Hospital Virgen del Camino de Pamplona. Navarra. Disponible en http://www.seom.org/seomcms/images/stories/recursos/sociosyprofs/documentacion/socios/2006/retinoblastoma/guiaRetinoblastoma.pdf

7. Alonso A, Moreno S. GUÍA DE MANEJO DE RETINOBLASTOMA [en linea] Hospital Virgen del Camino de Pamplona. Navarra. Disponible en http://www.seom.org/seomcms/images/stories/recursos/sociosyprofs/documentacion/socios/2006/retinoblastoma/guiaRetinoblastoma.pdf

Grupo B3
Subgrupo 3
NOMBRE
|
CARNE
|
Mayra Alejandra Sandoval
Rosa
|
201210311
|
José Manuel Alvizures
Pérez
|
201400185
|
Idalis Gavina Pérez Tomás
|
201400309
|
Camila Orellana Zepeda
|
201400349
|
Samantha Noribelly Ortíz
Chojolán
|
201407627
|
